In this case study, we hear from Dr Alan Jones (Pharmacy), who has been making use of BlueBEAR to enable his research in drug discovery.
My name is Alan Jones. I am an Associate Professor based in the School of Pharmacy. My research interests are linked to drug discovery in the widest sense (making, optimising, and developing new medicines).
For several years, our research group has been interested in how a class of sulfated molecules, called glycomimetics, have a beneficial effect on experimental models of cardiovascular disease. Our particular interest has been on atherosclerosis, a wide-ranging spectrum of disease states beginning with endothelial dysfunction and terminating with vascular calcification.

As part of a drug discovery project, we have used more routine desktop protein-ligand docking to guide and inform the development of novel glycomimetic analogues and how they interact with our principal target, hepatocyte growth factor (HGF). HGF/c-Met signalling affects vascular calcification with heparan sulfate acting as the native ligand in the body. Downstream signalling of HGF/c-Met modulates the P13K/Akt pathway leading to the production of nitric oxide a biomarker of endothelial function.
The docking model was built using the X-ray crystal structure of NK1-HFG (PBD: 1BHT) and each glycomimetic was docked individually, and the minimum energy of interaction calculated. Using this computational approach, we were able to rank and then triage which key molecules to put forward for additional biological screening. With the potential key interactions determined via docking studies we have synthesised glycomimetics with interesting structure-activity effects on nitric oxide levels.
The high-performance computing power required to understand the real-time movement of both protein and small molecule for a short time frame, for example, 50 nanoseconds, required the supercomputing power of BlueBEAR
Critically for our studies, we were able to perform molecular dynamics simulation (alongside MM/PBSA calculations) using the BlueBEAR facility at the University of Birmingham. Routine molecular docking using commercial software such as AutoDock can be performed on a laptop but the high-performance computing power required to understand the real-time movement of both protein and small molecule for a short time frame, for example, 50 nanoseconds, required the supercomputing power of BlueBEAR.
The dual use of MM/PBSA calculations and molecular dynamics allowed us to obtain a more accurate prediction of binding free energy of the most promising glycomimetics. By using BlueBEAR we were able to incorporate conformational fluctuations, entropic contributions, and solvent molecules interactions, not considered by molecular docking that is based on simple energetic and geometric criteria. Exploration of detailed energy composition obtained from MM/PBSA, revealed that the predominant interaction of a negatively charged glycomimetic toward the N-domain of the protein is electrostatic in nature.
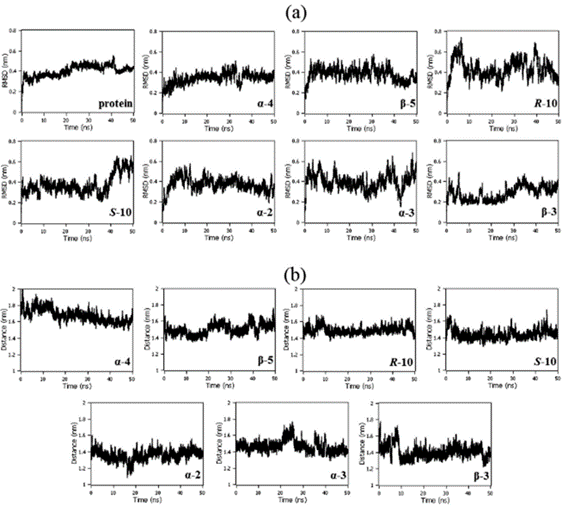
Root-mean-squared deviation (RMSD) analyses showed the stability of the protein structure along 50 ns of molecular dynamics in the presence of each glycomimetic. Using BlueBEAR we found that the glycomimetics did not cause any abrupt movements in the protein, indicating that the protein remained stable in the presence of the glycomimetic. The protein remained stable after 10 ns of simulation in the presence of the molecules with small fluctuations (Figure 1a). The distance between the centres of geometry (COG) of the N-domain with each glycomimetic structure remained stable with small fluctuations, indicating that the molecules remained in the binding site along 50 ns of molecular dynamics (Figure 1b). This has given key insight into our continuing interest in the design and optimisation of glycomimetics for cardiovascular indications.
Reference
Gill, D. M., Povinelli, A. P. R., Zazeri, G., Mahmoud, A. M., Shamir, S. A., Wilkinson, F. L., Alexander, M. Y., Cornelio, M. L., Jones, A. M. “The modulatory role of sulfated and non-sulfated small molecule heparan sulfate-glycomimetics in endothelial dysfunction: Absolute structural clarification, molecular docking and simulated dynamics, SAR analyses and ADME studies” RSC Med. Chem. 2021, 12, 779-790.
https://pubs.rsc.org/en/content/articlelanding/2021/md/d0md00366b
We were so pleased to hear of how Alan and his colleagues are able to make use of what is on offer from Advanced Research Computing, particularly to hear of how he has made use of Compute on BLueBEAR– if you have any examples of how it has helped your research then do get in contact with us at bearinfo@contacts.bham.ac.uk. We are always looking for good examples of use of High Performance Computing to nominate for HPC Wire Awards – see our recent winners for more details.