In this case study we hear from Philippa Cox, a PhD student in Chemistry, who has been using BlueBEAR to characterise novel materials for renewable energy applications.

I am a first year PhD student in the Scanlon Materials Theory Group based at the University of Birmingham. My research centres around using computational methods to analyse the structure, electronic properties, and defect chemistry of emerging energy materials.
It is fairly common knowledge amongst researchers that the world needs to drastically change its approach to energy generation in order to reduce the environmental impact of meeting the worlds’ growing energy demand. Although this problem ultimately needs to be approached from a range of directions, one of the most powerful tools we have is the ability to screen a large number of materials using high performance computing. This allows us to find cheaper, more abundant, and environmentally kinder alternatives, whilst also improving their performance. In the Scanlon Materials Theory Group, this ranges across a huge number of applications such as batteries, photovoltaics (solar cells), thermoelectrics (heat to energy conversion), transparent conductors (transparent electrodes), photoelectrochemical (water splitting) and many others.
My personal interest is in the field of thermoelectrics and photovoltaics, looking at how the symmetry and disorder that can exist within a materials structure impacts the electronic properties and the conversion efficiency. I look at both ordered and disordered materials. For ordered materials, we take the repeating unit of a material (known as a unit cell), create what is known as a supercell by putting a few of these unit cells together, and then we run our calculations using a method called Density Functional Theory. This allows us to study the properties of this system with relatively low computational cost. For disordered systems it is a little more complicated, we end up having to employ techniques such as cluster expansions and Monte Carlo integration methods.
This is work that would simply not be feasible on standard desktops.
To demonstrate the importance of high-performance computing in our work, consider that the radius of an atom is typically in the range of 10-10 meters. Now consider that for each atom there is a set number of electrons (a radius of approximately 10-18 meters) defined by the type of atom and the surrounding environment. These electrons contribute significantly to the properties and bonding in this material. If we were to then imagine how many atoms and electrons are in, for example, 1 cm3 of pure silicon, the number would be too impractically massive to model accurately enough to analyse the electronic properties.
For our purposes, each interaction between electrons and atoms in the material needs to be quantified as accurately as possible. For example, even a slight change in bond length can impact the electronic properties to a degree where a material previously considered useful is no longer worth pursuing. Density Functional Theory allows us to get high accuracy results but requires a huge number of computations to do so. To demonstrate the cost of Density Functional Theory, in 2011 Yukihiro Hasegawa’s group in Japan used 6144 cores of the K supercomputer to calculate the electronic states in a silicon nanowire of 107292 atoms, this took approximately 24 hours1.
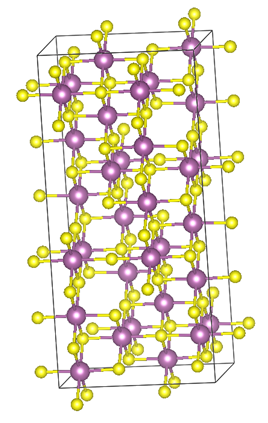
Currently, I am looking at a couple of ordered systems that have unit cells ranging from 5 to 80 atoms and hundreds of disordered systems that have supercells that may contain up to 192 atoms. One of these calculations on BlueBEAR can take anything from between a couple of hours to many days to run, depending on the accuracy and properties required, and I will have done hundreds of thousands of calculations by the time I finish my PhD. This is work that would simply not be feasible on standard desktops.
To find out more about the work going on in the Scanlon Materials Theory Group head to Scanlon Materials Theory Group | Computationally Driven Materials Design (davidscanlon.com) .

We were so pleased to hear of how Philippa was able to make use of what is on offer from Advanced Research Computing, particularly to hear of how she has made use of BlueBEAR HPC and its many cores – if you have any examples of how it has helped your research then do get in contact with us at bearinfo@contacts.bham.ac.uk. We are always looking for good examples of use of High Performance Computing to nominate for HPC Wire Awards – see our recent winners for more details.
1 Y. Hasegawa, J.-I. Iwata, M. Tsuji, D. Takahashi, A. Oshiyama, K. Minami, T. Boku, F. Shoji, A. Uno, M. Kurokawa, H. Inoue, I. Miyoshi and M. Yokokawa, in Proceedings of 2011 International Conference for High Performance Computing, Networking, Storage and Analysis, Association for Computing Machinery, New York, NY, USA, 2011, pp. 1–11.